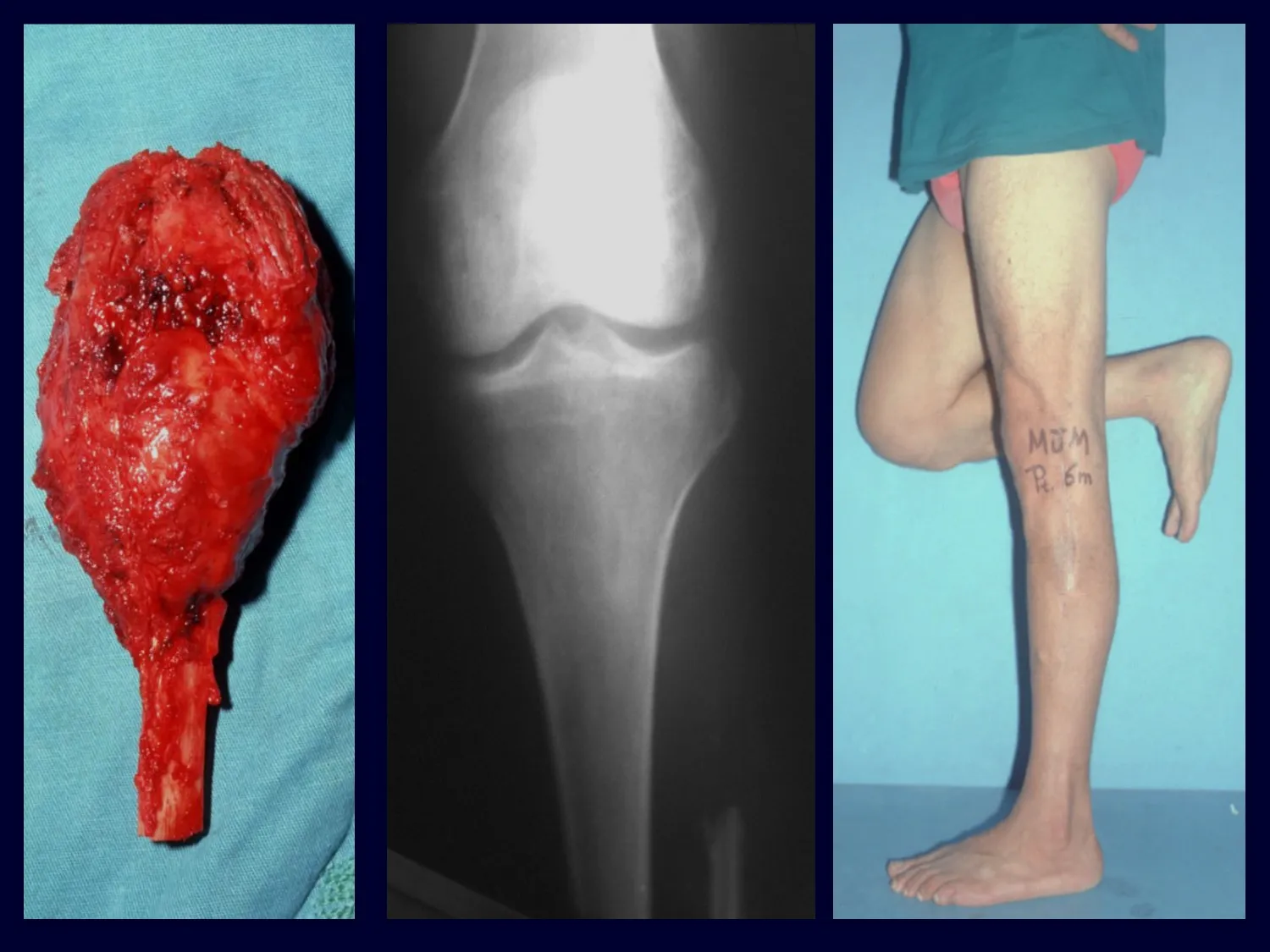
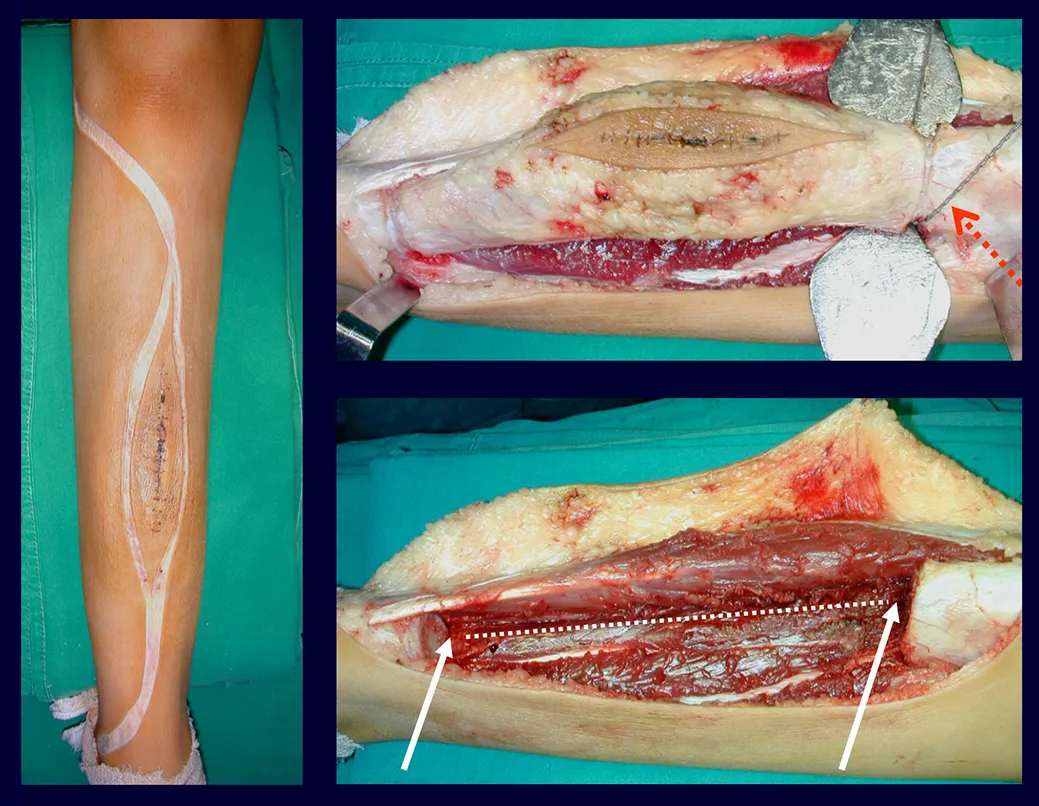
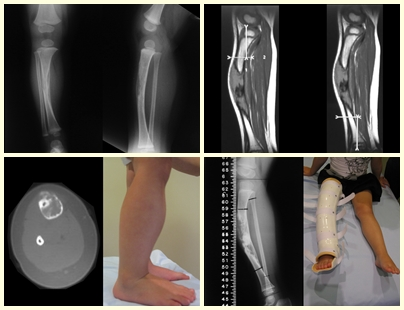
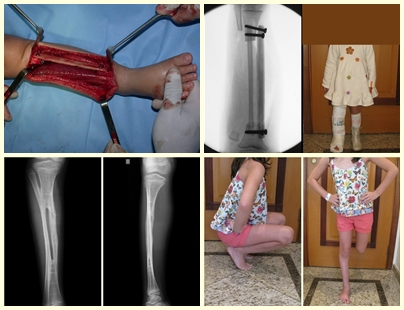
In single lesions, treatment is curettage and bone grafting when necessary.
Definition: Non-neoplastic lesion of unknown etiology, characterized by an intense proliferation of reticulohistiocytic elements with a variable number of eosinophils, neutrophils, lymphocytes, plasma cells and multinucleated giant cells. Frequent areas of necrosis, as well as the presence of fat cells, especially in old and multiple lesions.
Incidence: Reticuloendotheliosis presents several forms of involvement, but is mainly divided into three basic forms: Eosinophilic Granuloma (75%), Hand-Schuller-Christian (15%) and Letterer-Siwe (10%).
Eosinophilic granuloma: 5 to 20 years
Hand-Schuller-Christian: 3 to 5 years
Letterer-Siwe: 1 to 3 years
Etiology: Reticuloendotheliosis does not have a known etiology, however some authors relate it to a probable viral or immunological cause, due to the presence of an inflammatory phenomenon with the formation of a hyperplastic granulomatous process, often similar to neoplastic processes.
Clinical Manifestations: The natural history of the evolution of this disease will depend on one of the three forms in which it presents itself.
– Eosinophilic granuloma: most of the time it presents as a single lesion, preferentially affecting the diaphyseal and metaphyseal region of the long bones, and more rarely we also see cases with multiple involvement, which can be simultaneous or consecutive, starting in adolescence and dragging itself into young adulthood. Single injuries often end up resolving spontaneously over time, ranging from months to years, and are rarely disabling or lead to a pathological fracture.
– Hand-Schuller-Christian: normally presents with multiple lesions, which are more difficult to treat and evolve in a more disabling way than Eosinophilic Granuloma. They frequently present secondary involvement of other tissues, frequently progressing to Diabetes insipidus (involvement of the parapituitary gland), exophthalmos due to orbital involvement and involvement of the liver and spleen.
– Letterer-Siwe: the most frequent clinical findings are fever, otitis media and a frequent history of bacterial infections, and in some cases there is anemia, hepatosplenomegaly, bleeding with no apparent cause, lymphadenopathy and disseminated bone lesions. Evolution is often fatal due to systemic involvement.
Radiographic Aspects: The lesions have a radio-transparent appearance, with a rounded and ovoid shape, with well-defined and well-defined edges, and trabeculae within them can often be visible. They frequently affect the diaphyseal region of long bones and less commonly in the metaphyseal region, causing cortical erosion and slight cortical expansion. It is possible to visualize a small periosteal lift with an “onion skin” reaction similar to that of Ewing Sarcoma and osteomyelitis.
When the involvement is in the spine, it rarely leads to neurological impairment, although there is a collapse of the vertebra, presenting a flattening and known as “Calvé’s flat vertebra”.
In more serious cases, such as Hand-Schüller-Christian Syndrome and Letterer-Siwe Syndrome, disseminated radio-transparent lesions are observed in the cranial vault.
Treatment and Prognosis : The treatment and prognosis of the disease depend directly on the degree of involvement and clinical manifestations. In single lesions, the treatment of choice is curettage and in large defects, filling with cancellous bone. In some cases where there is no impairment of function or aesthetic impairment, resection of the compromised bone can be performed, such as the ribs, clavicle, and upper part of the fibula. In cases of multiple and systemic involvement, part of the treatment is carried out with the use of chemotherapy drugs and corticosteroid therapy.
1- Click to see more: http://bit.ly/granuloma_eosinoófilo-por
2- Case of polyostotic eosinophilic granuloma : http://bit.ly/Granuloma_Eosinófilo_do-Rádio
Cortical fibrous defect / Non-ossifying fibroma
The cortical fibrous defect is a benign non-neoplastic bone lesion, of unknown cause, which is characterized by fibrous proliferation in a small area of cortical bone. Non-ossifying fibroma is the same process, with a larger size.
The cortical fibrous defect generally does not present any clinical symptoms or signs. In the vast majority of cases it is diagnosed in an x-ray examination carried out for some reason. When it takes on the characteristics of a non-ossifying fibroma, it may manifest as mild pain, a protrusion noticeable by the patient or, less frequently, a fracture.