Giant Cell Tumor
Characteristics, Diagnosis and Treatment
Giant cell tumor, also known as TGC, is a mesenchymal neoplasm that is notable for the proliferation of large multinucleated cells, called gigantocytes. These cells resemble osteoclasts and are found within a stroma of mononucleated cells. Due to its peculiar histological morphology, accurate diagnosis often requires a thorough analysis of the clinical and radiographic picture in order to avoid confusion with other pathological processes.
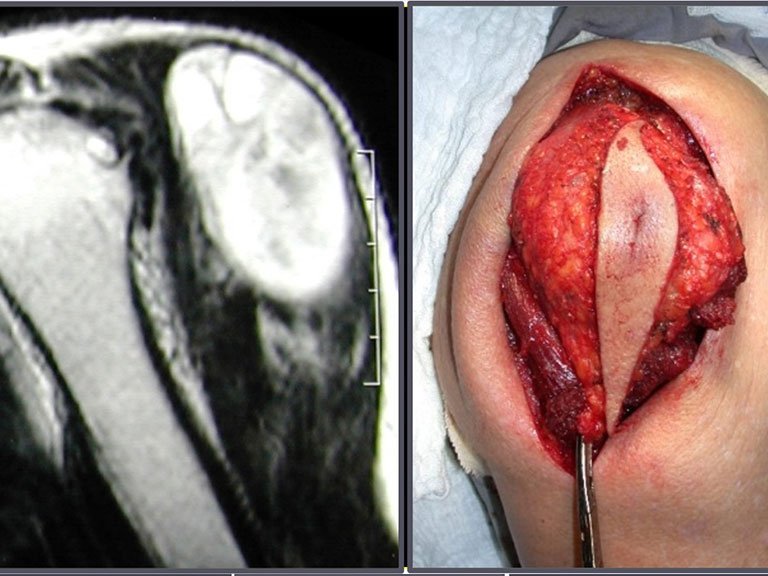
The main manifestation of a giant cell tumor is intermittent local pain, often accompanied by an increase in volume in the affected region and restriction of adjacent joint movements. The evolution period varies from 6 to 12 months, depending on the affected bone, with reports of trauma as the initial trigger of symptoms being common.
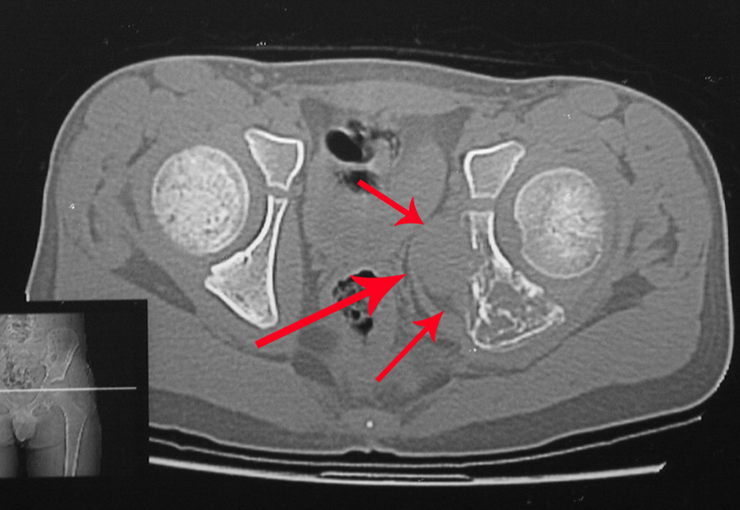
This type of tumor tends to affect a single bone, mainly long bones such as the femur, tibia, humerus and radius. However, in rarer cases, it can occur in bones of the axial skeleton, with a predilection for the sacrum. The incidence is more common between the third and fourth decades of life, affecting both sexes equally.
Radiographically, TGC appears as an epiphyseal lesion characterized by bone rarefaction, initially eccentric and later compromising the cortex. Diagnostic confirmation is obtained through histological analysis, which reveals the presence of multinucleated giant cells and spindle cell stroma.
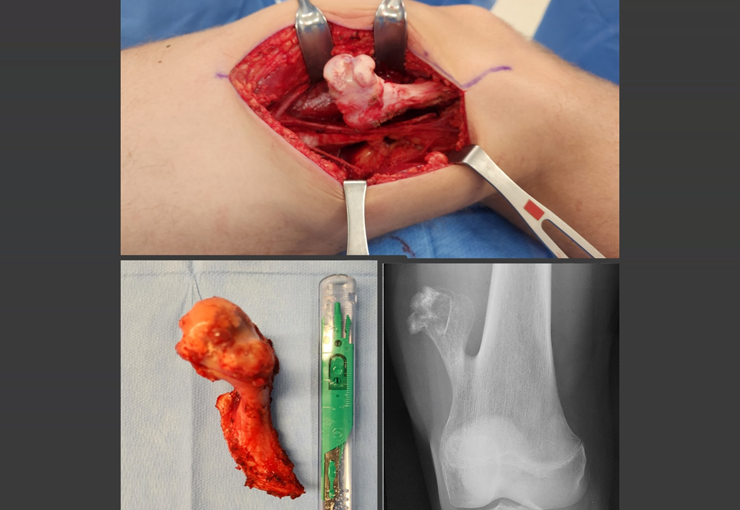
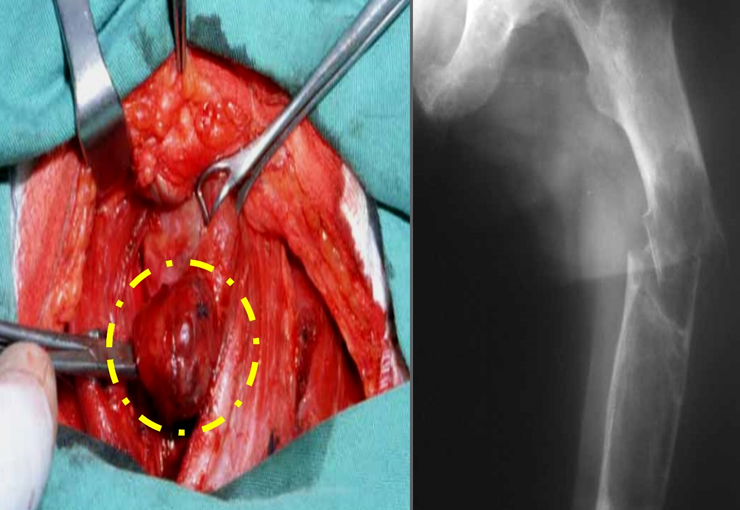
The treatment of giant cell tumors is well established and aims at segmental resection of the lesion, whenever possible, ensuring safety margins for both bone and soft tissues. In cases where segmental resection is not feasible, such as in the cervical spine, endocavitary curettage followed by adjuvant therapy is indicated. Among the adjuvant therapeutic options are the CO2 laser, phenol diluted in 4% alcohol, liquid nitrogen and electrothermia.
The electrothermal technique has been shown to be effective in complementing curettage, providing a more complete cleaning of the tumor cavity. After electrothermia, milling of the cavity using appropriate instruments such as the slow drill is performed to ensure complete removal of the remaining tumor cells.
Filling the treated cavity can be done with different materials, such as autologous bone graft, bone substitutes or methylmethacrylate, each with its advantages and disadvantages. Post-treatment follow-up is essential to monitor disease recurrence and ensure the effectiveness of the treatment performed.
In summary, giant cell tumor is a complex condition that requires a multidisciplinary approach to ensure the best therapeutic outcome and the patient’s quality of life. Advances in diagnostic and treatment techniques have contributed significantly to improving the prognosis of these patients, offering increasingly effective and safe therapeutic options.
Author: Prof. Dr. Pedro Péricles Ribeiro Baptista
Orthopedic Oncosurgery at the Dr. Arnaldo Vieira de Carvalho Cancer Institute
Office : Rua General Jardim, 846 – Cj 41 – Cep: 01223-010 Higienópolis São Paulo – SP
Phone: +55 11 3231-4638 Cell:+55 11 99863-5577 Email: drpprb@gmail.com