Classification:
The World Health Organization (WHO) classifies soft tissue sarcoma according to the histological subtype that the neoplasm resembles, such as liposarcoma, synovial sarcoma, rhabdomyosarcoma, etc. 1
In some cases, the histology is uncertain and the morphological aspect is used to classify them as alveolar sarcoma or clear cell sarcoma.
The most common soft tissue sarcomas in adults are: undifferentiated, liposarcoma, synovial sarcoma, leiomyosarcoma and malignant tumor of the peripheral nerve sheath, which is included in this chapter, despite originating in the ectoderm, as it presents biological behavior , treatment and prognosis similar to soft tissue sarcomas 1,2 .
Histological grade is also used to classify soft tissue sarcomas, being classically divided into Grade 1 , well differentiated with a low degree of histological malignancy; Grade 2 , moderately differentiated and Grade 3 , poorly differentiated with a high degree of malignancy 1 .
Clinical condition:
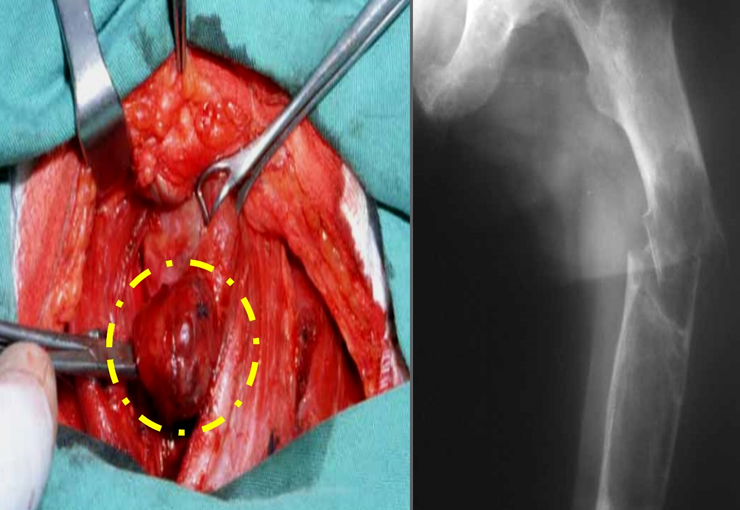
The initial clinical picture is palpable tumor bulging, often painless, with progressive growth that can reach considerable size, especially in the thigh.
Some patients may eventually experience pain and paresthesia due to the compressive effect of the tumor. They are clinically eutrophic in most cases, with fever or weight loss being exceptional symptoms.
Soft tissue sarcomas tend to grow between adjacent muscles, pushing and compressing the surrounding structures, rarely extending beyond the anatomical compartments. Growth speed varies between histological subtypes.
There are no absolute clinical criteria to differentiate benign from malignant soft tissue tumors. However, all deep tumors, that is, below the plane of the muscular fascia, and superficial tumors greater than 5 cm, have a high probability of being malignant 2 .
Staging:
At the time of diagnosis, soft tissue sarcoma rarely metastasizes, not exceeding 10 percent of cases. This occurs more frequently in large volume tumors, deep to the muscular fascia and of high grade 2 .
The dissemination pattern for most sarcomas is hematogenous and the main site of metastasis is the lung. For this reason, all patients with soft tissue sarcoma must undergo chest staging. Chest tomography to search for lung metastasis should always be indicated, especially for high-risk patients, such as superficial lesions larger than 5 cm, deep tumors and those with a high degree of malignancy.
Regional lymph nodes are the site of metastases in 2.6% of cases, however, in some histological subtypes these may be more frequent and should be investigated, such as rhabdomyosarcoma, synovial sarcoma, epithelioid sarcoma, clear cell sarcoma and angiosarcoma 2 .
Tomography of the abdomen and pelvis is recommended, especially in the staging of round cell soft tissue sarcoma and myxoid liposarcoma, due to the high rate of extrapulmonary metastasis to the abdomen and retroperitoneum. In the case of angiosarcoma, evaluation of the central nervous system, through skull tomography, is necessary due to the propensity for metastasis to the brain.
Imaging exams:
a) Radiography:
Radiography is the cheapest and most accessible imaging test. It can be used as the first line of tumor evaluation, to differentiate a tumor of skeletal origin from a soft tissue sarcoma. Furthermore, it may show calcifications inside.
b) Magnetic resonance imaging:
Magnetic resonance imaging is still the test of choice to evaluate soft tissue sarcomas when there are no metallic elements, such as an orthopedic prosthesis. It can detail the limits of the tumor and its relationship with neighboring structures. The use of contrast can also differentiate solid lesions from lesions filled with liquid, and the examination using contrast should be requested.
c) Tomography:
Tomography is the exam of choice in retroperitoneal tumors and in the search for pulmonary metastasis, as resonance is not adequate due to respiratory movement and intestinal peristalsis. In the study of MTS of extremities, resonance is superior in delineating neoplastic extension 2 .
d) Pet-Cet:
Fluorine deoxyglucose positron emission tomography (FDG-PET/CT) allows, with high sensitivity, to differentiate high-grade soft tissue sarcoma from benign soft tissue tumors. However, it loses value in differentiating between low-grade and low-grade tumors. intermediaries 2 .
FDG-PET/CET should not be used routinely in the initial evaluation of a patient with tissue tumor, however its use is indicated in determining prognosis and response to chemotherapy treatment 2 , 3 .
The standaruptakevalue (SUV) is a quantification of fluorine-labeled glucose consumed by the tumor and demonstrates tumor metabolism. Soft tissue sarcoma with SUV equal to or greater than 6, which after chemotherapy shows a reduction in this value of less than 40 percent, presents a higher risk of systemic recurrence of the disease 2 .
e) Scintigraphy:
Bone scintigraphy is not always performed in the initial staging of soft tissue sarcoma, as bone metastases in adults are infrequent in the initial stage of the disease. The exception is round cell tumors and myxoid liposarcoma, but skeletal mapping can be false negative and evaluation using magnetic resonance imaging is recommended 2 .
Biopsy:
Biopsy is indicated for the histological diagnosis of MTS, since imaging tests do not present characteristics suggestive of the histological subtype, as we can infer in some bone neoplasms, such as osteosarcoma.
A biopsy is not always necessary for treatment, as the treatment of MTS is still primarily surgical and, in some cases, the biopsy can be replaced by excisional resection or also called excisional biopsy.
Superficial tumors, above the muscular fascia, smaller than 5 cm, have a low probability of being malignant. In these cases, they can be resected without histological diagnosis if they are subject to oncological surgery (wide resection with an oncological margin, without compromising surrounding noble structures) , as this would be the correct treatment in the event that we are dealing with a high-quality soft tissue sarcoma. degree.
The biopsy must be performed by the surgeon who will carry out the definitive treatment or be guided by him. The biopsy path must be in line with the incision that will be used for resection and this entire biopsy path must be removed en bloc, along with the tumor.
Incisional biopsy, performed through a surgical incision, exposing part of the tumor, was the most used for the diagnosis of soft tissue sarcoma. Currently, percutaneous biopsies have gained ground with the development of appropriate needles, such as the Trucut®, and imaging methods such as ultrasound and tomography to guide them.
Pathology:
The pathologist must always be present to perform the frozen section examination, in order to confirm and ensure that the material collected is representative of the lesion, avoiding the need to repeat the procedure. The definitive histological result must await paraffin histology and possibly immunohistochemistry.
Percutaneous needle biopsy, performed with these precautions, generally makes it possible to differentiate malignant tumors from benign ones and experienced pathologists are able to correctly determine the histological grade of the tumors in 97.6 and 86.3 percent respectively, in addition to this technique causing less local dissemination of the tumor. injury during the procedure 3 .
Soft tissue sarcoma – Treatment:
The treatment of soft tissue tumors is generally surgical, however, as with most neoplasms, monitoring of cancer patients must always be multidisciplinary.
From diagnosis to staging and treatment, a team of several professionals interacts in managing the case, such as the orthopedist who will perform the oncological surgery , the pathologist, the radiologist, the clinical oncologist, the radiotherapist, the psychologist, the social worker, etc.
The objective of treatment is to preserve the patient’s life, avoiding local recurrence, maximizing the function of the affected limb and minimizing treatment morbidity.
Chemotherapy:
Soft tissue sarcomas, in the pediatric population, respond to systemic chemotherapy by presenting a greater response to drugs, providing an improvement in survival, as the child is able to withstand the doses of chemotherapy necessary to control the disease, unlike adults.
In children, the most common soft tissue sarcomas are rhabdomyosarcoma, the extraosseous form of osteosarcoma, and the extraosseous form of Ewing’s sarcoma. All of these with neoadjuvant and adjuvant chemotherapy protocols.
In the rare cases of these neoplasms occurring in adults, the pediatric chemotherapy protocol is chosen, at doses supported for adults and their possible comorbidities.
In adult soft tissue sarcomas, there is great controversy regarding the use of chemotherapy. The meta-analysis published in 1997 by the Sarcoma Meta-analysiscollaboration showed that the use of doxorubicin in these patients increased the time free from local recurrence and distant metastasis, but there was no statistically significant benefit in the patients’ overall survival 3 .
A new updated meta-analysis was published in 2008 demonstrating that the use of doxorubicin associated with ifosfamide promoted an improvement in patient survival 3 . However, chemotherapy is extremely toxic, especially in the adult population where the presence of clinical comorbidities can make effective treatment unfeasible. The current recommendation is to individualize each case, remembering that systemic treatment does not correct an inadequate surgical resection.
In general, adult MTS are poor responders to chemotherapy. Treatment, therefore, relies on surgical resection.
Oncological surgery:
Soft tissue sarcoma resection surgery must be performed with wide margins, so that the entire tumor is removed en bloc and covered by healthy tissue, aiming to ensure that there are no residual neoplastic cells after resection.
Soft tissue sarcomas have a surrounding pseudocapsule. This structure is represented by an inflammatory process produced by tumor aggression and is not always a sufficient barrier for neoplastic cells, which permeate the surrounding tissues. Adjuvant radiotherapy may be indicated to act on the margins when they need to be small, due to the need to preserve noble structures, such as vessels and nerves.
Tumor resection through the pseudocapsule can leave microscopic neoplastic tissue in the patient and may be a risk factor for local recurrence and worse prognosis.
Soft tissue sarcomas tend to grow by expanding and pushing against surrounding tissue, but rarely by infiltrating it. Therefore, invasion of bone or adjacent muscle tissues is uncommon.
Most of the time when the tumor approaches the bone, it is possible to dissect it by removing the periosteum that surrounds the bone in question, along with the tumor. This deperiostization associated with radiotherapy, which is generally used as adjuvant, increases the risk of fracture, especially in the femur 4 .
Recurrence:
The thickness of the wide margins around the tumor is questionable, with 1 cm being recommended, but this rarely happens homogeneously throughout its circumference.
To preserve noble tissues, such as large nerves or vessels, the surgeon eventually reduces this margin to avoid resection of such a structure, which can put both the patient and the limb itself at risk in order to preserve the best function.
On the other hand, anatomical tissue planes represent different barriers to the tumor, such as muscular fascia where 1 to 2 millimeter margins may be adequate for safe resection.
Other tissues such as fat or muscle belly require wider margins.
Large nerves can be preserved by dissecting and removing the outer nerve sheath as a margin, since soft tissue sarcomas do not usually infiltrate nerves. When the tumor surrounds the nerve, there is greater technical difficulty and this nerve may need to be sectioned and reconstructed with microsurgical sutures.
Nerve grafts for reconstruction are rarely useful in the lower limbs of adults, especially in a site that will need to be irradiated. Children, however, may have better results.
Ablative surgery is generally avoided, as even limited function of the lower limb may be viable, even in cases requiring complete resection of the sciatic nerve.
Radiotherapy:
The use of adjuvant radiotherapy promotes the destruction of tumor cells around the main lesion. When associated with limb-preserving surgical resection, it promotes increased local control, minimizing recurrence to 10 to 15% 4 . However, the association of this therapeutic modality does not increase overall survival.
Radiotherapy is indicated for almost all cases of soft tissue sarcoma.
Exclusive surgical resection may be sufficient for cases of low-grade superficial tumors or small-volume, totally intramuscular tumors, as long as wide margins are guaranteed in the oncological surgery performed.
With the improvement in local control, radiotherapy as an adjuvant in soft tissue sarcomas has reduced the rate of amputation of extremities from 50% in the 1970s to 1% today, without compromising survival 5 .
The indication for amputation in the presence of MTS may be necessary in excessively extensive tumors, such that the residual limb has no function.
An example is the need to resect a main nerve trunk, such as the brachial plexus, with the impossibility of obtaining wide margins. In this case, the loss of sensation results in a non-viable remaining limb.
The involvement of large vessels by the tumor was already an indication for amputation in the past, however with the vascular reconstruction technique it became possible to resect the tumor en bloc with the vessels and reconstruct them with a vascular graft, allowing a viable limb.
Surgical margins:
The main variable in local control of the disease is the surgical margins of tumor resection 5 .
The presence of margins coinciding with the tumor increases the risk of local recurrence and recurrence increases the risk of distant metastasis, which can lead to shorter survival.
The presence of compromised margins, however, does not represent certainty of local recurrence, since only 30 percent of cases undergoing incomplete resection and undergoing adjuvant radiotherapy suffered recurrence.
Wide margins, on the other hand, are also no guarantee of local control, since, even when associated with radiotherapy, recurrence in this situation is around 5 to 10 percent.
The presence of positive margins in the resection may require a new approach to the surgical bed, seeking to expand the previously obtained margins.
This new approach will depend on the analysis of which anatomical structure will need to be resected for this expansion.
In reference centers for oncological surgery, the presence of compromised margins is usually related to a possible residual tumor in large nerve bundles that would require amputation for resection. If this is the case, a higher dose of radiotherapy (66-68 Gy) can be used or amputation can be chosen as a last resort.
Benign soft tissue tumors:
Benign soft tissue tumors are one hundred times more common than malignant tumors. Among them, subcutaneous lipoma is among the most diagnosed in clinical practice.
These lesions do not cause pain and are diagnosed when they are superficial and protrude into the skin or when they are deep and reach a large volume. The exception is schwannoma that arises from a peripheral nerve and can present pain, paresthesia and, less frequently, motor changes.
Despite being benign, as these tumors grow, compression of adjacent structures may occur and only then will symptoms appear.
The treatment of most benign soft tissue tumors depends on the symptoms they eventually produce. As most of them are asymptomatic, surgical resection is indicated when they increase in size, causing discomfort or aesthetic changes.
In superficial, small-sized, asymptomatic tumors without signs suggestive of malignancy on imaging exams, it is possible to choose to observe the clinical evolution.
Desmoid tumor, despite being benign, is a locally aggressive soft tissue neoplasm, which produces symptoms by attacking neighboring tissues.
Unlike most benign soft tissue neoplasms, desmoid resection should be indicated with wide margins, an oncological surgery similar to the treatment for malignant tumors.
Desmoid tumor is highly recurrent, even after adequate resection. In some cases, the patient may require amputation of the limb due to numerous recurrences, or when experiencing intractable pain or dysfunction and a new resection becomes impossible.
Discussion:
- One consideration is to perform a prior biopsy or excisional biopsy ( biopsy resection ) in cases of small, superficial soft tissue sarcomas, or in places where oncological surgery is possible, with wide margins around the entire circumference of the tumor?
- Another consideration is regarding adult soft tissue sarcomas, in which neoadjuvant chemotherapy does not improve locally nor interfere with survival. There is controversy regarding the use of neoadjuvant radiotherapy:
- A) Radiotherapy + surgery + radiotherapy or
- B) Surgery + radiotherapy?
Table 2 below summarizes parameters that we must consider.